分子对接案例
《项目方案评估》
1. 蛋白序列
CRT:蛋白质晶体结构RCSB PDBID:3POS
A VYFKEQFLDG DGWTSRWIES KHKSDFGKFV LSSGKFYGDE EKDKGLQTSQ DARFYALSAS FEPFSNKGQT LVVQFTVKHE QNIDCGGGYV KLFPNSLDQT DMHGDSEYNI MFGPDICGPG TKKVHVIFNY KGKNVLINKD IRCKDDEFTH LYLIVRPDN TYEVKIDNSQ VES
IGG:蛋白晶体结构2DLF、1WZQ、1GLC,此处挑选2DLF
Structure Obtain The amino acid sequence of CRT and IGG were retrieved from the sequence database of NCBI (www.ncbi.nlm.nih. gov). The three-dimensional structures of CRT and IGG were both available in Protein Data Bank (PDB ID: 3POS, 2DLF). |
2. 对接位点分析
根据文献报道并结合模板蛋白晶体结构,进行蛋白质相互作用预估分析,整理其可能相互作用的结合模式。
在以下文献中对该蛋白所属6类亚型蛋白进行了序列比对,发现该蛋白序列特别是binding domain存在着极高的序列相似性,高达95%以上,说明该蛋白结合槽位置高度保守,这一推论表明我们所研究的蛋白质在“结构组成”和“结合模式”上都是较为统一。
因此,该文献具有相对较为可靠的参考价值,参照本文献可以进行相似方法的蛋白分析。

3. 蛋白蛋白对接
结合已有相关资料与蛋白对接软件Rossetta,对蛋白质多聚体进行构建。
Ø 蛋白蛋白对接一直是分子模拟中非常重要同时非常难解决的话题,相较于小分子蛋白之间的联系,蛋白蛋白对接如今更加不成熟,在蛋白蛋白对接之前,最好能够搜集更多的文献进行支持,让模拟的结果不空洞,才能保证对接的准确性。
蛋白蛋白对接如今比较著名的软件有Hex Protein Docking,ZDock,rDock,以及Rosetta等等。其中Hex Protein Docking虽算法比较复杂,但没有评分功能(PS:可能是我没有找到···),ZDock一般作为前期的初对接,rDock为ZDock的升级算法,一般将ZDock对接后的前几个得分构象使用rDock进行进一步对接。但是Hex Protein Docking,ZDock和rDock对接方法,目前均处于低级对接阶段,对接结果基本不可信,特别是本文中的大体系跨膜蛋白分子对接项目。
Rosetta对接软件,是由加州大学旧金山分校著名晶体结构物理学家David Baker及其项目组所开发研究,进展使接近原子精度的蛋白质预测和设计成为可能。这一软件,基于高精度高计算效率的全原子能量函数和用于搜索极端崎岖势能面的有效采样策略的开发,这两者都由结构预测和设计的测试结果的反馈所推动。Rosetta程序中统一的能量和运动框架可广泛用于大分子建模的问题中,从微纤结构预测到RNA折叠再到设计新的蛋白质界面,都易于进行研究和突出区域的改进设计。其对接结果相较于Hex Protein Docking,ZDock和rDock等几个软件,有着极大的提升与可靠性。
Rosetta中进行对接包含两个步骤。第一步,进行积极(aggresive)采样,方法使用的质心模型。第二步采用全原子模型进行小范围的优化。
Rosetta软件最早是在华盛顿大学David Baker教授实验室开发的,目前软件内有多个应用可供用户使用,常用的应用程序有同源建模(comparative modelling)、短片段模拟与重建(Loop modelling and rebuilding)、蛋白质设计(protein design)、蛋白质与蛋白质对接(Protein-protein docking)、蛋白质配体对接(Protein-ligand docking)等。Rosetta软件对于学术界用户是免费的,只需要申请获得一个许可证,就可以从Rosetta的官网中下载软件了。
Protein-Protein Docking A geometry-based molecular docking software called ROSETTA was used for docking the predicted three dimensional models of CRT and crystal structure of IG. The software predicts the docked transformations that produce good molecular shape complementarity. The algorithm divides the Connolly dot surface representation of the molecules into concave, convex, and flat patches. The patches were matched according to their complementarities in order to generate different transformations. A default value of 4 A˚ was used for clustering and redundant solutions were discarded by RMSD clustering. The Patch Dock output generates the geometric score, desolvation energy, interface area size, and the actual rigid transformation of the solutions. Twenty solutions, out of 60 predicted complexes, were sorted according to their geometric shape were again refined through the fire dock server. The complementarities scores were analyzed for identifying the residues involved in the protein-protein interface. |
3.1 Rosetta对接结果挑选
利用Rosetta软件共对接出10000中蛋白相互作用位相。利用Rosetta自带聚类分析模块进行聚类分析并根据分子对接能量打分进行排名。其中位置比较靠谱,能量打分较好的前10位蛋白质对接能量如下: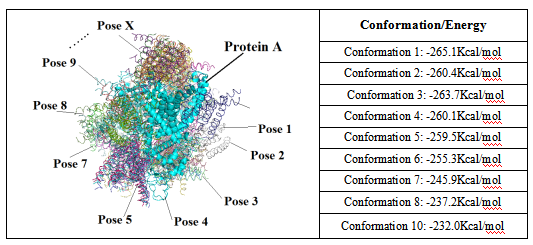
我们取其中能量最好的结构Conformation 1 (-265.1Kcal/mol)进行后续分析。
4. 蛋白分子动力学优化
Molecular Dynamics Simulation Molecular dynamics (MD) simulations were performed using the GROMACS 4.5 package [67]. The GROMACS program package (http://www. gromacs.org) adopting the OPLSAA force field parameters were used for EM and MD simulations. The protein-protein complex were analyzed in three separate system to obtain the stable conformation of the protein and protein-protein complex for analyzing three dynamic behavior of these structures. The bad contacts from the 3D structure of the proteins were refined and solvated with the solvent [68]. The system was further relaxed by energy minimization and for the MD simulation studies, the structures were solvated using the TIP3P water model and the solvated structures were energy minimized using the steepest descent method, terminating when maximum force was found smaller than 100 KJ/mol-1/nm-1. All the simulations were performed in the NPT ensemble at constant temperature (300 K) and pressure (1 bar) with a time step of 2 fs. NVT were performed for 1ns (nanoseconds) and the minimized structure were equilibrated with timescale of 10 ns (nanoseconds). Trajectory conversion and RMSD scripts were used for analysis of MD simulation. |
经过结构叠合与补齐,并对构象蛋白质进行6ns分子动力学优化,直至结构平衡。
6.1 方法
选取最优构象,用AMBER14软件做分子动力学模拟。整个蛋白体系都采用gaff和ff14SB力场,以蛋白为中心,加10A的立方水盒子,加Na+使体系呈电中性,保存拓扑和坐标结构,然后进行模拟,模拟流程如下:
(1)两步能量最小化,先限制蛋白,最小化水分子的能量,然后放开蛋白,整个体系的能量最小化。第一次能量最小化一共5000次循环,先采用最速下降法做1500次循环,第二次能量最小化一共5000次循环,先采用最速下降法做了2000次循环。
(2)体系平衡,首先是体系的升温平衡过程,采用郎之万控温方法平衡了100ps。 然后进行升压平衡过程,一共平衡了100ps,采用了各向同性的 Berendsen 控压方法。
(3)动力学模拟,无限制自由模拟阶段。控温控压方法与前一阶段相同,范德华能和短程静电能的截断距离为10Å,采用PME方法计算长程静电能。
6.2 结果分析
6.2.1 RMSD值分析
对该蛋白进行4次分子动力学优化,从四个体系骨架原子的均方根波动值(RMSD值)可以看出,在2ns后,四个体系RMSD值的波动都小于0.5A,均已经达到平衡状态,可以用来结构分析。说明该蛋白质,无论始发点出于什么状态,初始参数均能使蛋白质得到相应的结构优化,并达到较好的平均结构状态。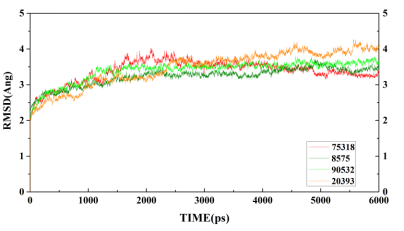
4次体系随时间变化的RMSD值
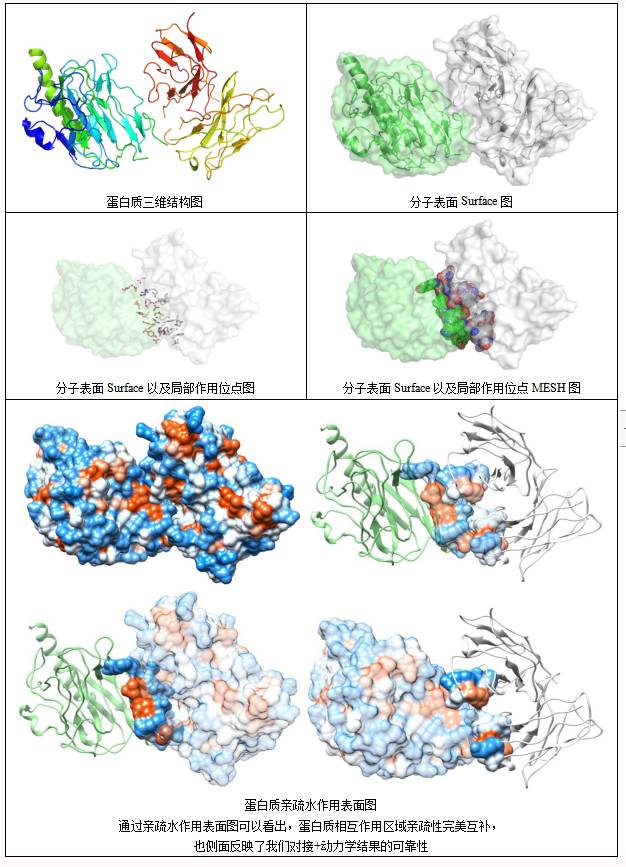
取出平均结构进行后续分析,得到的平均结构如下:
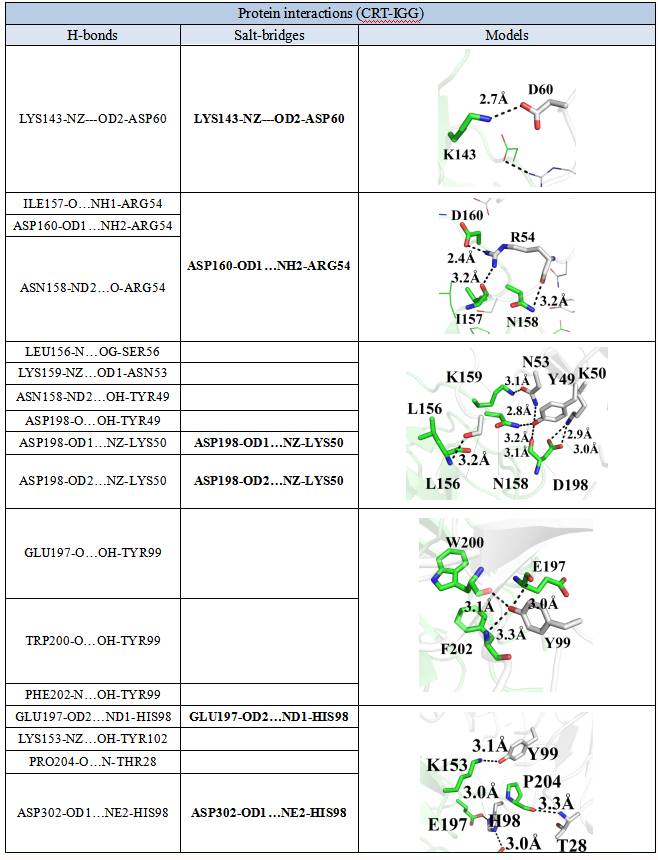
Protein interactions (CRT-IGG) | |||
H-bonds | Pi-Pi | Salt-bridges | disulphide bonds |
ASN53-ND2…OH-TYR49 | None | None | |
LYS143-NZ---OD2-ASP60 | LYS143-NZ---OD2-ASP60 | ||
LYS153-NZ…OH-TYR102 | |||
LEU156-N…OG-SER56 | |||
ILE157-O…NH1-ARG54 | |||
ASN158-ND2…O-ARG54 | |||
ASN158-ND2…OH-TYR49 | |||
LYS159-NZ…OD1-ASN53 | |||
ASP160-OD1…NH2-ARG54 | ASP160-OD1…NH2-ARG54 | ||
GLU197-O…OH-TYR99 | |||
GLU197-OD2…ND1-HIS98 | GLU197-OD2…ND1-HIS98 | ||
ASP198-O…OH-TYR49 | |||
ASP198-OD1…NZ-LYS50 | ASP198-OD1…NZ-LYS50 | ||
ASP198-OD2…NZ-LYS50 | ASP198-OD2…NZ-LYS50 | ||
TRP200-O…OH-TYR99 | |||
PHE202-N…OH-TYR99 | |||
PRO204-O…N-THR28 | |||
ASP302-OD1…NE2-HIS98 | ASP302-OD1…NE2-HIS98 | ||
